Home
Home
-
National Metabolomics Data Repository
Upload and Manage
StudiesBrowse and Search
StudiesAnalyze
StudiesAs of 01/18/26 a total of 4458 studies have been processed by the National Metabolomics Data Repository (NMDR). There are 4006 publicly available studies and the remainder (452) will be made available subject to their embargo dates.
Recently released studies on NMDR
ST004537 - C8 positive-mode LC–MS lipidomics of BMDM and PMN: WT vs KO (n=22); Mus musculus; Harvard Medical School
ST004500 - Benchmarking Data for Untargeted Mass Spectrometry-Based Metabolomics: LC (Reverse Phase)-Orbitrap MS (Negative); Homo sapiens; Washington University in St. Louis
ST004501 - Benchmarking Data for Untargeted Mass Spectrometry-Based Metabolomics: LC (HILIC)-Orbitrap MS (Positive); Homo sapiens; Washington University in St. Louis
-
New RefMet molecule page layout
RefMet molecule page
The new RefMet molecule page layout now contains distinct sections covering nomenclature, molecular descriptors,chemical/biochemical classification, distribution information in NMDR studies (including species and tissue plots), external identifiers to other databases and MS/NMR spectra of standards where applicable. See an example here.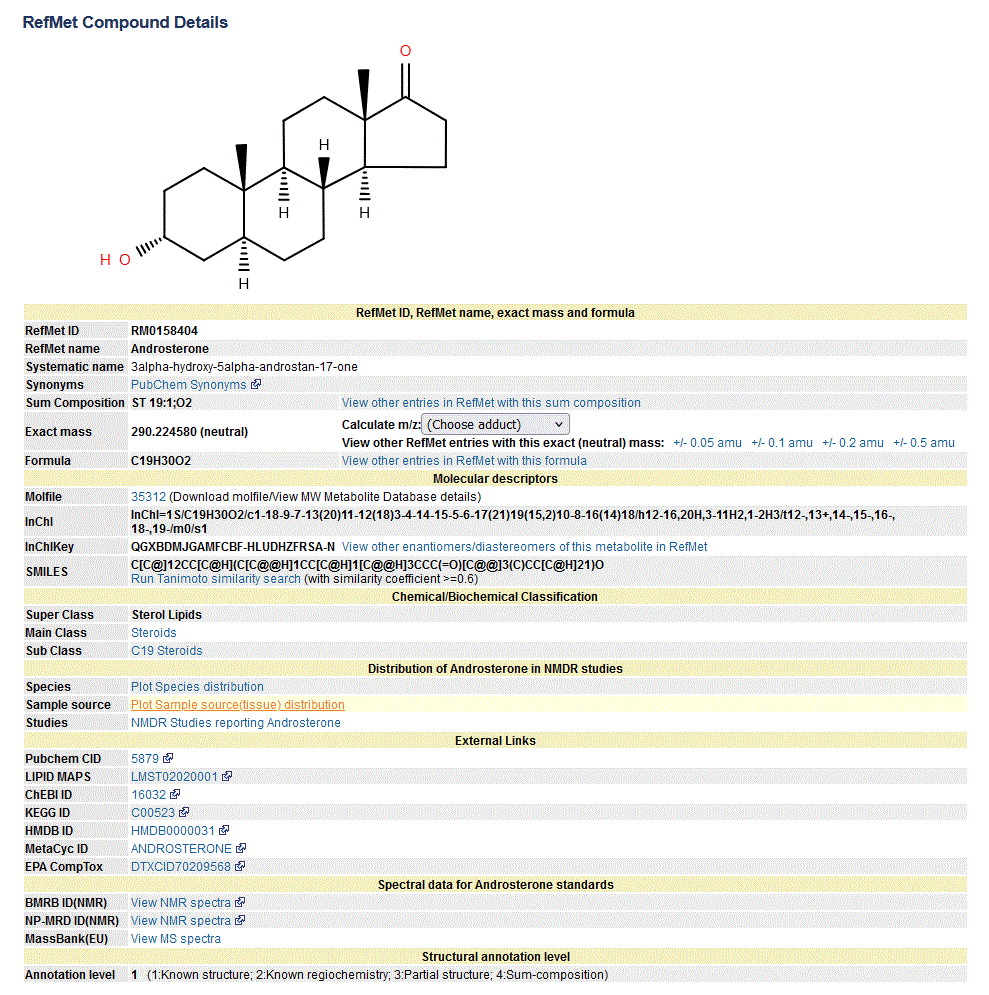
-
Focus on sterol drawing tools and nomenclature
Sterol drawing tools
This web-based sterol drawing tool contains a selection of 45 different cores (including bile acid amidates for proteogenic amino acids) and numerous functional group options. It generates downloadable structures in molfile or image formats, as well as systematic name, formula and exact mass, with an option to calculate m/z values for various ion-adducts. This tools is also available on LIPID MAPS. There is a new table of bile acid nomenclature covering RefMet names, systematic names and abbreviations.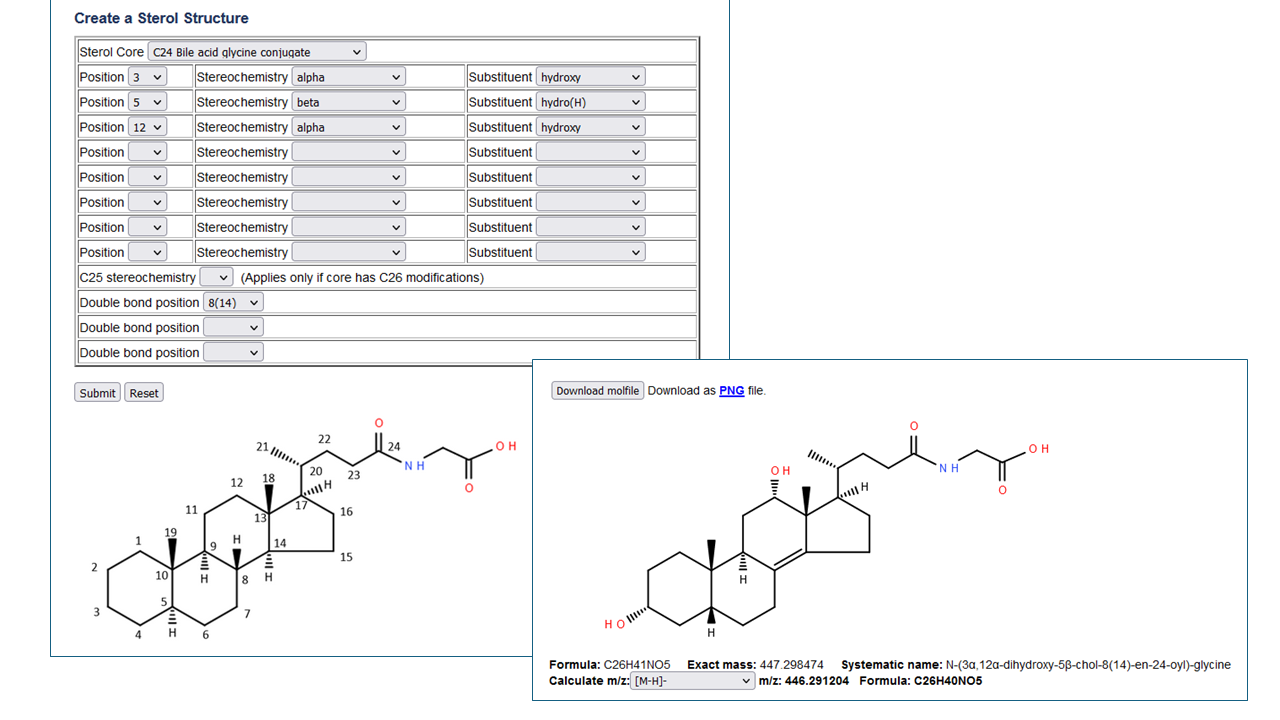
-
RefMet name harmonization combined with ion-adduct calculation
RefMet ion-adduct calculation
The RefMet metabolite name harmonization resource leverages data from over 500,000 annotations obtained from MS and NMR studies in NMDR to provide a standardized reference nomenclature across 4 different levels of structural resolution. A new feature is the ability to map a list of metabolite names to RefMet and simultaneously perform an ion-adduct calculation to generate a list of m/z values.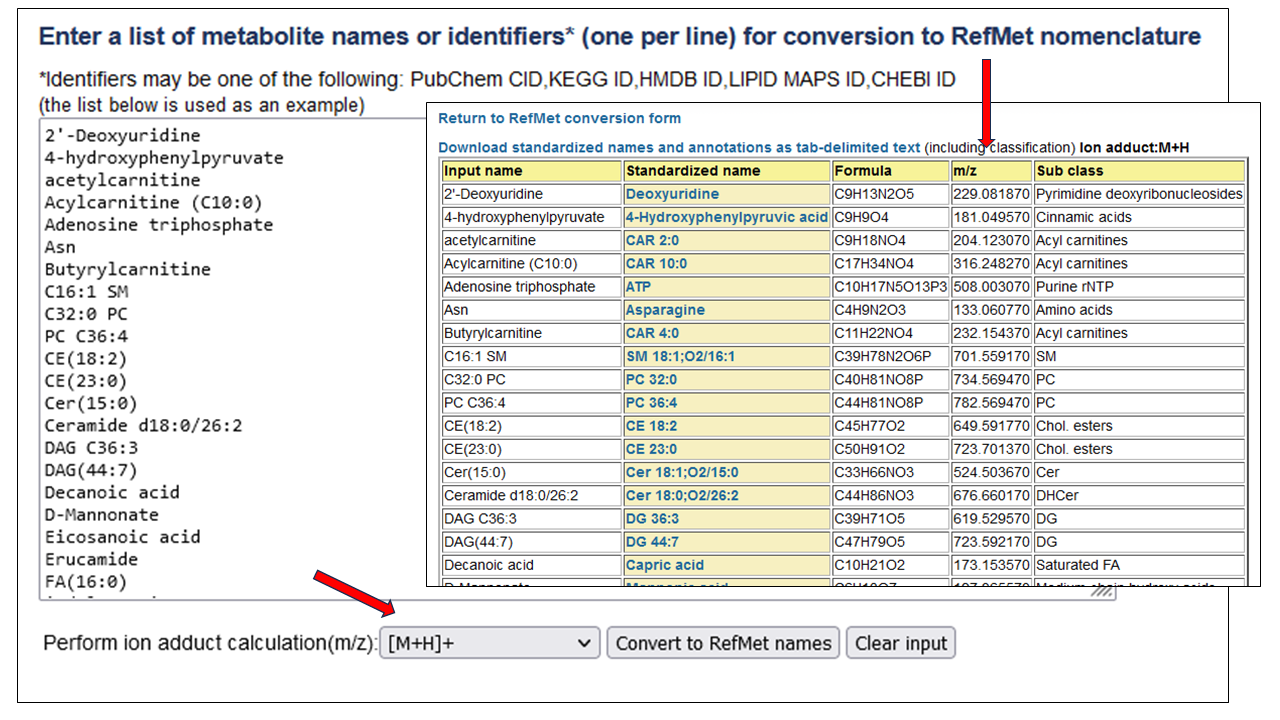
-
Core structures in RefMet classification
RefMet core structures
Browse and search core structures associated with the RefMet classification system. For example, what are "Flavones", "Flavanols", "Flavonols" and "Flavanones" and what's the difference? The RefMet classification hierarchy has recently been updated to place more emphasis on biosynthetic considerations for Alkaloid, Polyketide and Prenol lipid super classes.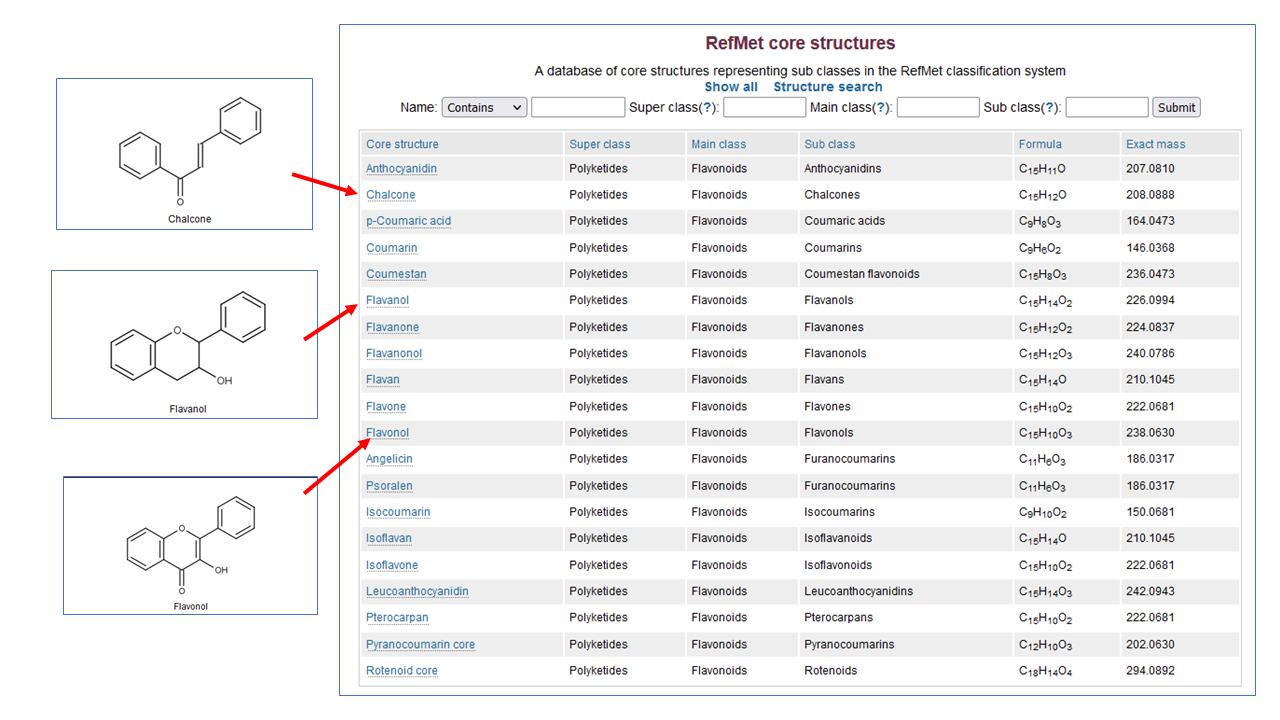
-
Molecular structure similarity analysis
Create molecular structure similarity networks
This Structure similarity network tool creates a network map from a list of metabolite names (up to 500) by selecting a fingerprint type (MACCSkeys, Chem.RDK, Topological, Morgan,MorganBitVector) and similarity method (Tanimoto, Dice) with a similarity coefficient cutoff. This feature is also implemented for each NMDR study containing named metabolites (in 'Perform statisctical analysis' section). This application uses the Python-based Rdkit.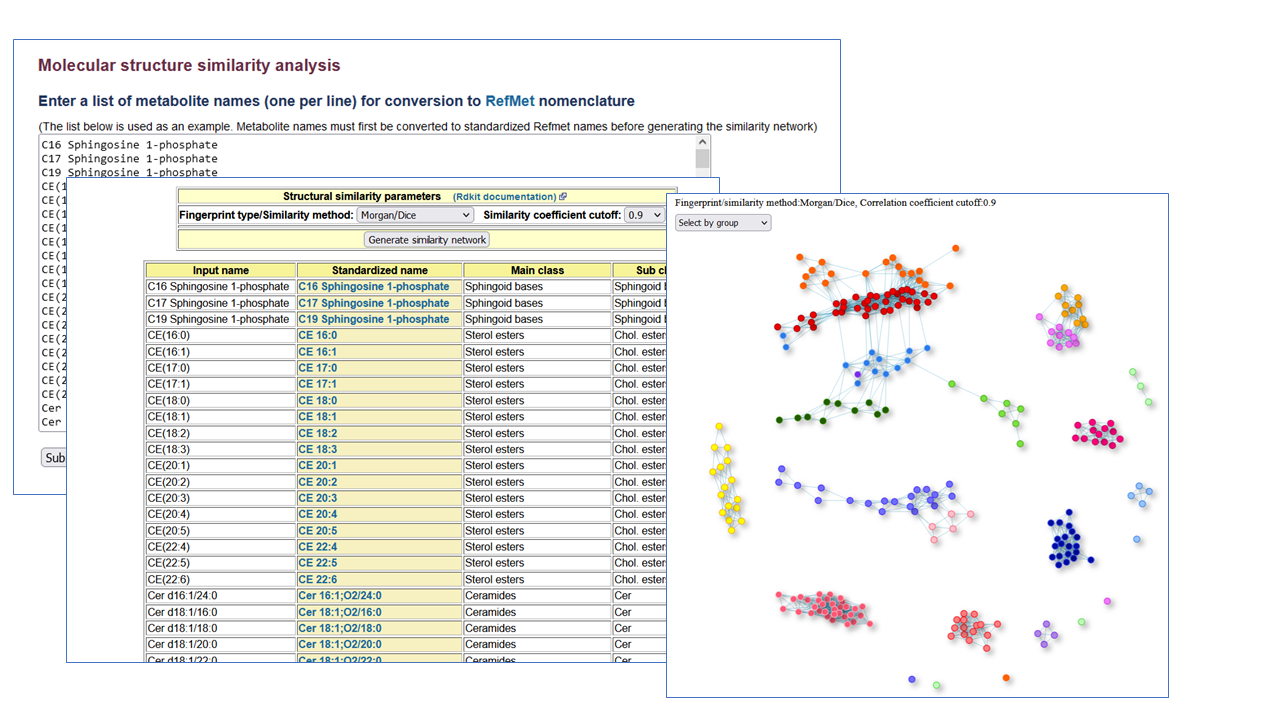
-
Searching untargeted LC-MS data
Searching untargeted LC-MS data on the Workbench
This portal searches over 4.5 million m/z,retention time features from over 890 NMDR studies and over 1500 LC-MS analyses. Search with a m/z value and tolerance window and optionally specify a retention time value and tolerance window to restrict the search. Limit search to studies by sample source and/or species, and also by chromatography type, MS instrument and polarity. Features that have been identified by submitters will appear in the "Name" column in the results table.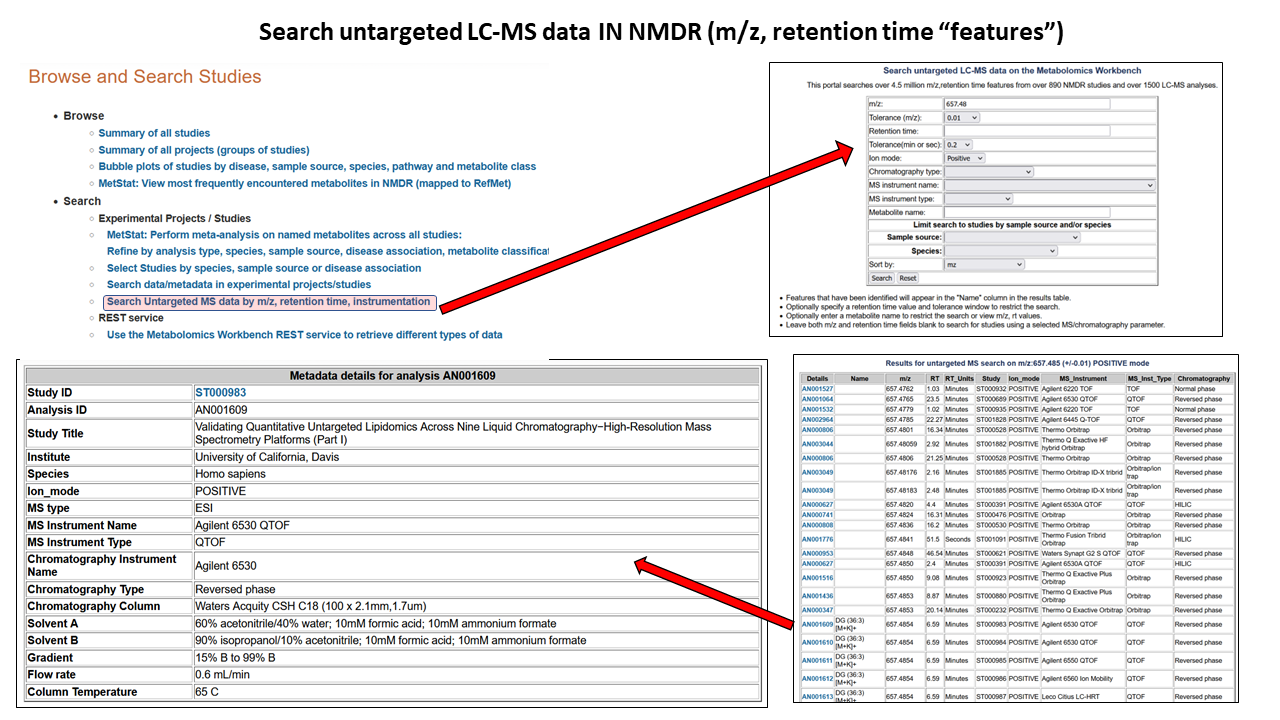
-
Correlated network graphs in NMDR
Correlated network graphs using Debiased Sparse Partial Correlation (DSPC)
The Metabolomics Workbench has released a new graphical tool for estimating and visualizing partial correlation networks in NMDR studies. It uses the Debiased Sparse Partial Correlation algorithm (DSPC) developed at U.Michigan. Nodes may be mapped to chemical classification or fold-change. Study example: See "Perform Network analysis on correlated metabolites" links here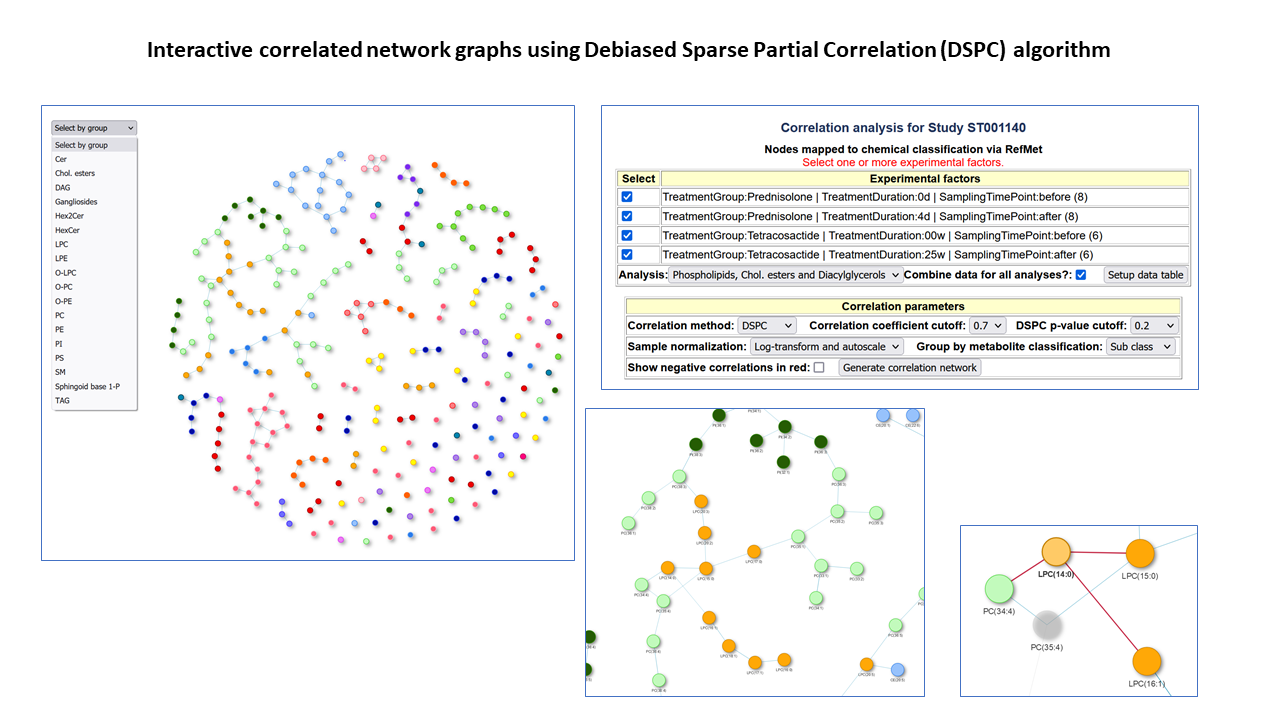
-
Lipid Notation in RefMet and lipid m/z calculation tools
View table of over 170 revised lipid abbreviations covered by RefMet, including structure examples and m/z calculation tools for a variety of adducts.
-
Convert your metabolite name to standardized nomenclature via RefMet
-
Exemplary Studies
A list of exemplary studies are listed here which adhere to the submission guidelines of Metabolomics Workbench. Specifically, publically available studies having all or most of the features below were identified as exemplary studies.
- Well-written study summary
- Detailed metadata for collection/treatment/chromatography/MS/NMR, etc.
- Post-processing details
- Presence of control samples
- Raw data availability for samples and controls
- One-to-one mapping of sample names to raw data file name
- Internal standards (with measurements)
- Clear and organized metabolite annotations
These include different analysis (GC-MS, LC-MS, NMR) and species type. We recommend looking at these studies as a model example before submitting to Metabolomics Workbench.